








背景介绍
前言:细胞死亡被认为是导致炎症疾病的主要因素之一,此前已有很多证据表明阻断细胞死亡可以逆转炎症的病理状态。炎症细胞因子肿瘤坏死因子(TNF)是免疫反应中的重要细胞因子。它不仅直接驱动炎症基因表达,还间接地促使细胞死亡、参与炎症免疫反应和疾病发展。中和TNF在治疗慢性和急性炎症、自身免疫疾病中都获得了成功。因此,细胞死亡抑制剂被认为是治疗TNF依赖性炎症疾病的一种新疗法。
综述目的:通过回顾TNF作为治疗目标的历史和发现,介绍最近的研究结果,提高对各类细胞死亡程序、执行模式、检查点、病理和生理状态之间相互作用的认识,以此确认新的治疗靶点。
综述聚焦
一、TNF如何引起细胞死亡
起初研究者将阻断TNF受体后炎症反应减弱的效果归因于MAPK和NF-κB通路的激活减少,促炎症基因下调。即使这个认知可能是正确的,但更多研究表明TNF和其受体TNFR1可以通过诱导细胞凋亡、焦亡和坏死间接加剧炎症反应。
事实上,细胞的死亡并不是对TNF的默认反应,只有当一或多个检查点未激活时,TNF与TNFR1的结合才会激活细胞死亡程序(图1)。TNFR1通路如何进一步诱导细胞死亡的过程仍未知,但已确定它需要组装一个二级细胞膜复合体,称为“复合体II”。它可以结合和激活caspase 8,引起下游效应caspase以诱导细胞凋亡,或转而裂解gasdermin D (GSDMD)以触发细胞焦亡。
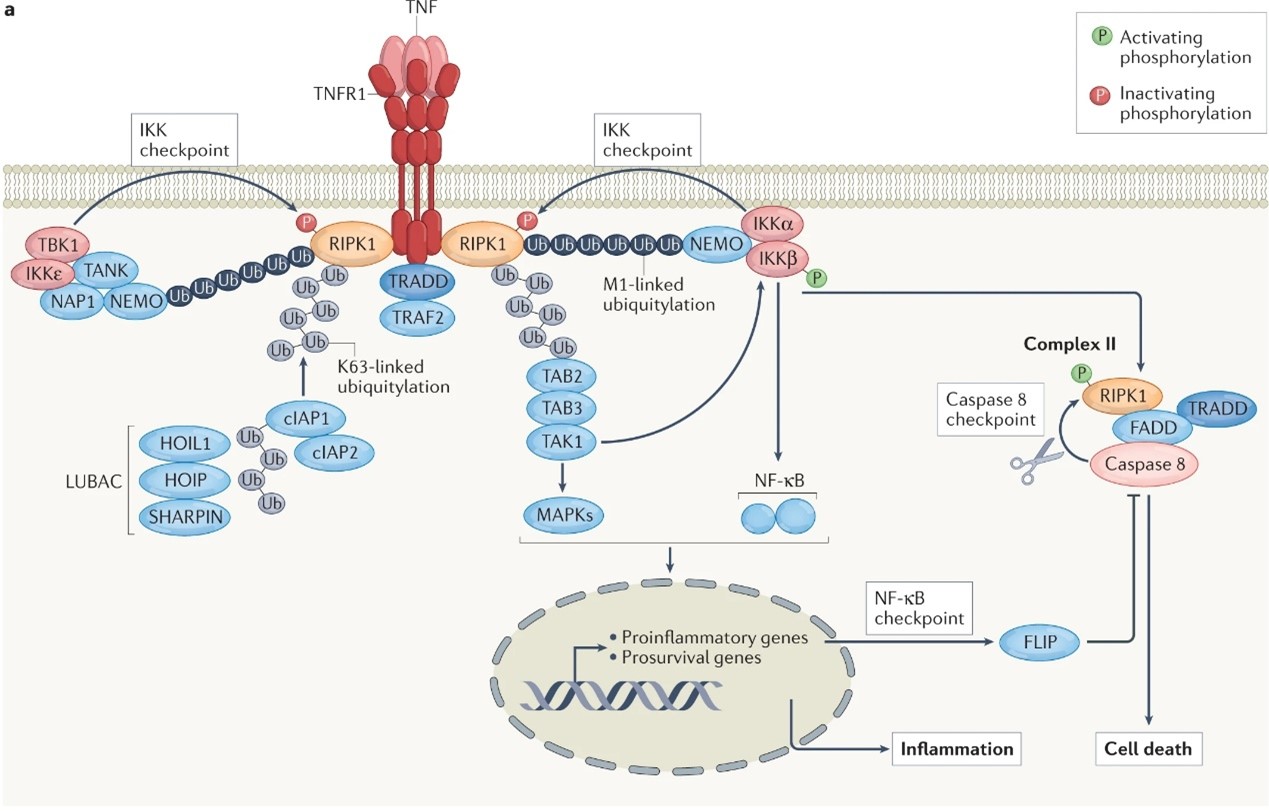
图1.TNFR1通路
当下已经有两种检查点被发现:IKK和NF-κB检查点(图2)。控制这两个检查点的激酶高度依赖泛素激活,因此与complex1的激活有关的抑制、突变或缺失都会激活TNF的细胞毒性。令人惊讶地是,即使上述的检查点都被抑制了,TNFR1的通路上依旧有第三个检查点通过裂解RIPK1激酶活性来恢复RIPK1的凋亡和坏死作用。
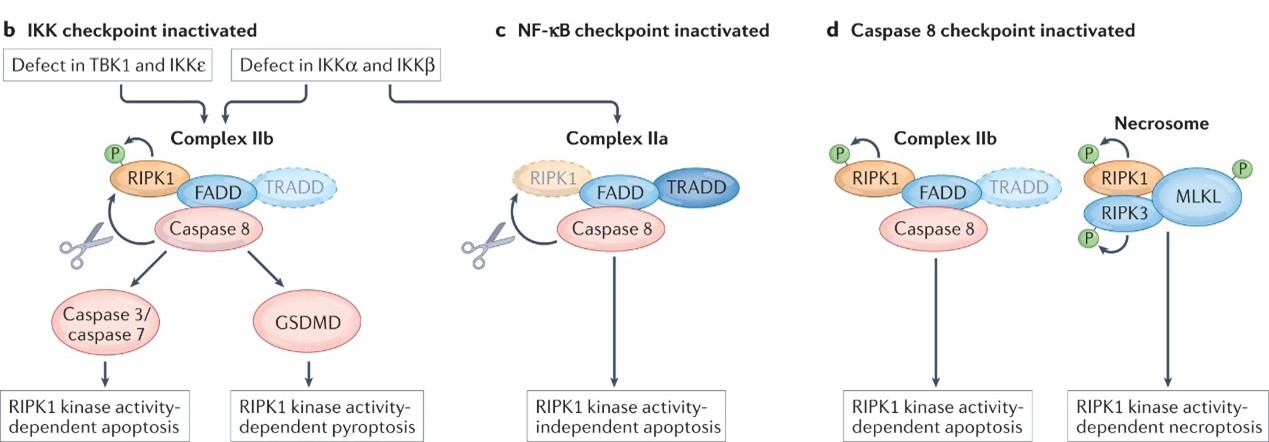
图2.三种检查点作用机制
二、TNF在病原体防御中诱导的细胞死亡
当宿主适应病原体的感染时,病原体也会随之发展新的机制来逃脱宿主的免疫系统攻击。病原体可以通过阻断其他的抗菌机制来劫持人体的炎症信号通路。不同的TNFR1细胞死亡检查点似乎是作为宿主对这种微生物的反应而进化而来的。当病毒想要通过释放效应分子或者毒素抑制炎症基因的激活时,细胞会改变自己的反应来诱导细胞死亡。这激活了另一种通过释放损伤相关的分子模式来提醒免疫系统的机制。细胞死亡因此被认为是一种宿主的防御机制。
TNF的细胞死亡并不总是对宿主有利。至少在某些特定情况下,TNF介导的细胞死亡的过度激活确实被证实为加剧而不是减少微生物的致病性和致命性,如在感染严重急性呼吸道综合征冠状病毒2(SARS-CoV-2)时观察到的那样。
三.TNF在炎症性疾病中诱导的细胞死亡
当异常诱导因环境因素或基因突变发生时,TNF诱导的细胞死亡会变成有害过程,成为各种(无菌)炎症性疾病的源头。多个研究已共同证明TNF在体内诱导RIPK1激酶活性依赖的细胞死亡,引起败血症休克。但这种情况只存在于体内模型,大多数细胞在体外不受单一的TNF刺激影响,其原因还不完全明确,一种合理猜测是细胞毒性可能源于多种细胞因子的共同感应。这些可能的附加配体包括TWEAK和淋巴毒素-β,在同族的配体和受体结合后,它们可以激活非经典的NF-κB通路。同样激活这条通路的还有TNF与TNFR2的结合,因此,TNFR1和TNFR2对TNF的共同感应也有可能将TNFR1的反应从生存转向RIPK1激酶活性依赖的死亡。
多项研究表明参与TNFR1途径的一些基因突变也会引起小鼠和人类的炎症性疾病:
- caspase8或FADD:缺乏这两种基因会导致胚胎死亡、炎症、IBD等等。
2.泛素信号调节基因:由于cIAP1、cIAP2和LUBAC与TNFR1复合物I共轭的泛素链间接控制抵消caspase 8依赖的细胞死亡的两个检查点,影响泛素信号正确调节的突变可以也引发TNF介导的异常细胞死亡。
- TBK1:作为先天免疫中的核心激酶,它在TNFR1通路中抑制RIPK1激酶活性。其缺失突变会导致人类早发的自体炎症综合征。
- FLIP: FLIP在NF-κB依赖的细胞存活中起着核心作用。由于卵黄囊血管发育的缺陷,Cflar(编码FLIP)的遗传性缺失导致胚胎死亡,FADD和Ripk3的联合缺失是防止FLIP缺陷小鼠致死表型的必要条件。
- RIPK1: 全身RIPK1消融导致出生后死亡,这被caspase 8和RIPK3的缺乏所拯救,表明RIPK1在抑制细胞死亡和随后的炎症方面有关键功能
原文链接:https://www.nature.com/articles/s41577-022-00792-3#Sec7








 沪公网安备31011502400759号
沪公网安备31011502400759号
 营业执照(三证合一)
营业执照(三证合一)




