








细胞的寿命可以从几天到很多年不等,具体取决于细胞类型。许多生理过程需要细胞死亡才能发挥其功能,例如,胚胎发育和 B 细胞和 T 细胞的免疫选择。在程序性细胞死亡(programmed cell death,PCD)的过程中,细胞核破裂成180-200 kD的DNA片段,内质网转变成囊泡,这些囊泡作为凋亡体释放到细胞外空间。含有细胞器和核碎片的凋亡体会被邻近细胞吞噬。
在感染,慢性组织损伤期间,当大量细胞突然死亡积累时,细胞死亡可能会导致内容物大量释放到细胞外空间。DAMPs(anger-associated molecular patterns)会被作为一种危险信号释放,引起一系列的免疫反应,招募胞吞和其他的免疫细胞,以此清除碎片和促进组织修复。
细胞凋亡,细胞坏死和细胞焦亡是最常被研究的三种程序性细胞死亡方式。此外,也有报道因为药物毒性引发的其他类型的程序性细胞死亡。
细胞凋亡
细胞凋亡很长一段时间被认为是唯一的程序性细胞死亡,并且是过去三十年的主要研究热点之一。它的关键特征之一是线粒体释放的细胞色素c,由BCL-2家族的促凋亡蛋白和抗凋亡蛋白、启动caspase(caspase-8、-9和-10)和效应caspase(caspase-3、-6和-7)之间的平衡调节。
从机理上讲,线粒体细胞色素c释放下游的caspase活化级联有两条主要途径:内在途径和外在途径。
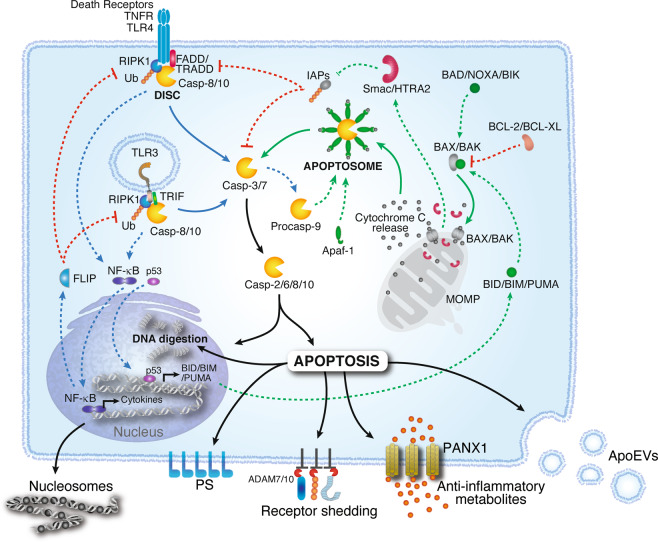
细胞凋亡通过两种主要途径触发,即内在途径和外在途径。内在途径是由BCL-2家族蛋白 BAK 和 BAX 的寡聚化激活的。BAK/BAX 寡聚体在线粒体外膜上形成孔,导致细胞色素 c 释放到细胞质中。BAK/BAX 的激活受促凋亡(如 BAD 和 BID)或抗凋亡(如 BCL-2)BCL-2 家族蛋白的调节。细胞色素 c 与 Apaf-1 结合,后者招募 procaspase-9,形成凋亡体。在凋亡小体中,caspase-9 通过自体蛋白水解作用被激活,从而启动了 caspase 处理级联。外源性途径通过膜受体(如TNFR1、TLRs)的参与而激活。这些蛋白会诱导形成信号复合体,其中包括TRADD或FADD、与受体相互作用的RIPK1和 procaspase-8。cIAPs对 RIPK1 的泛素化会稳定该复合物,并诱导转录因子 NFκB 的活化。同样存在于 DISC 中的 FLIP 可限制 caspase-8 的活性,同时促进细胞存活、细胞增殖和促炎细胞因子的产生。这一通路的失衡(如细胞应激所造成的失衡)会激活 caspase-8 和 caspase-10,进而触发 caspase 激活级联。一旦激活,执行级联caspase(即caspase-2、-6、-8和-10)就会导致程序性凋亡。凋亡细胞以核糖体结构、脱落受体、抗炎代谢物或凋亡细胞外囊泡分子的形式释放信使。暴露在质膜外表面的磷脂酰丝氨酸(PS)分子可作为吞噬细胞吞噬的信号
细胞坏死
这种类型的调控细胞死亡,一般由TNFR1的激活引起。其他的细胞受体也可以引起细胞坏死,包括Fas/FasL,TLR4,TLR3,以及RIG-I和STING,可以引起I型干扰素(IFN-I)和TNFα的产生,从而在自分泌反馈回路中促进细胞坏死。大部分的这些通路会促进NFκB依赖性促炎和促生存信号。微生物和药物对于蛋白酶Caspase-8的抑制也驱动了细胞坏死的通路。活性RIPK1被招募到包括FADD、caspase-8和caspase-10在内的寡聚复合物中。在Caspase-8活性被抑制的情况下,RIPK1招募和磷酸化RIPK3,形成一个叫做ripoptosome的复合体,接着招募和磷酸化MLKL,形成necrosome。MLKL的磷酸化引起了结构改变,4HBD结构的暴露。早期的研究猜测RIPK1/RIPK3/MLKL通路通过线粒体不稳化来引起细胞细胞死亡,DRP1依赖的和PGAM5依赖的通路主要参与了这个过程。之后这个假说被推翻,建立起另外两种模型:
1、MLKL是质膜上的一个平台,用于打开钙离子通道或钠离子通道,从而使离子流入、细胞膨胀和破裂;
2、MLKL自身在质膜上形成了孔洞,通过4HBD中带正电荷的补丁与膜上带负电荷的磷脂酰肌醇磷酸酯(PIPs)之间的相互作用。
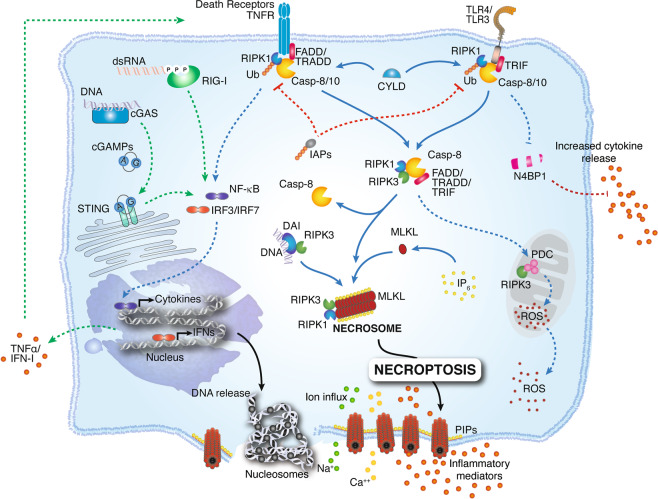
坏死性凋亡由死亡蛋白结构域(例如 TNFR 和 Fas)和 Toll 样受体 (TLR)-4 或 TLR3 的下游触发。激活后,这些受体会招募衔接蛋白 FADD、TRADD 和 TRIF,与 RIPK1 和 caspase-8 或 -10 相互作用。首先,RIPK1 被 IAP 泛素化,使其失去功能并通过 NFκB 实现促炎下游活性。检测到“死亡信号”后,RIPK1 被 CYLD 去泛素化,从而招募 RIPK3。RIPK1/RIP3 复合物招募并磷酸化 MLKL。在高度磷酸化的磷酸肌醇 (IP 6),磷酸化的 MLKL 寡聚化,从而形成坏死体。MLKL 寡聚物易位到质膜中富含磷脂酰肌醇磷酸 (PIP) 的斑块并形成大孔。最终,MLKL 孔通过允许离子流入、细胞肿胀和膜裂解以及随后细胞内物质不受控制的释放而导致坏死性细胞死亡。胞质核酸传感器 RIG-I 和 cGAS/STING 也有助于坏死性细胞死亡,因为它们诱导 IFN-I 和 TNFα,从而通过自分泌反馈环促进坏死性凋亡。在 TNFR 或 TLR 参与的下游,活性 caspase-8 会裂解细胞因子阻断剂 N4BP1,从而促进细胞因子释放的增加。一旦激活,RIPK3 就会磷酸化线粒体中的丙酮酸脱氢酶复合物 (PDC),并促进有氧呼吸和线粒体 ROS 产生。
细胞焦亡
细胞焦亡由炎症体传感器引起,最终会破坏质膜完整性,导致细胞死亡。这些炎症体传感器包括Nod样受体(NLR)家族、DNA受体Absent in Melanoma 2(AIM2)和Pyrin受体。他们可以检测各种PAMPs和DAMPs,通过细胞死亡阻止微生物的扩散。作为一把双刃剑,这种机制也可能会导致病理性炎症的发生。
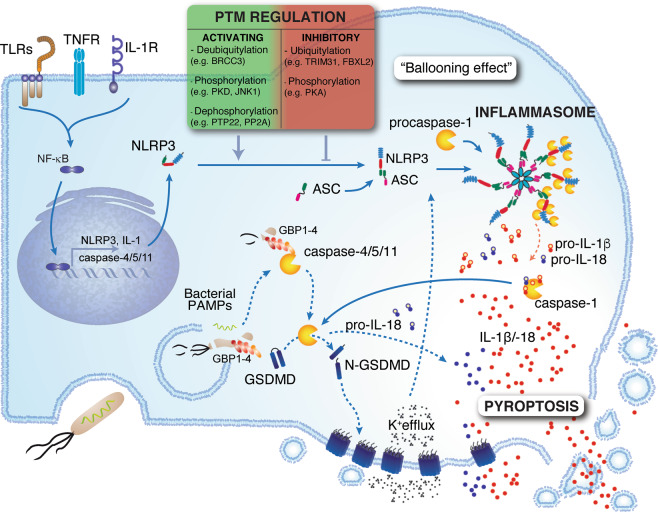
炎症反应是一种炎症性细胞死亡方式,主要由细胞内传感器触发,如NLRP3。激活后,这些传感器招募适配器ASC,形成一个微米大小的结构,称为炎性体。这些寡聚结构可以激活caspase-1,之后裂解白细胞介素-1家族细胞因子IL-1β和IL-18的原型。Caspase-1还能激活gasdermin D (GSDMD),解除N-末端结构域的屏蔽,从而在质膜上形成孔,进而使成熟的IL-1β和IL-18释放出来,引起细胞肿胀(气球效应)和细胞焦亡。
此外,细胞内对LPS的识别也可导致细胞裂解:鸟苷酸结合蛋白(GBPs)与细菌表面结合并组装一个由GBP1、2、3和4以及细胞膜前体caspase-4和-11组成的caspase-4/11激活平台。活跃的caspase-4和-11可裂解GSDMD,导致热凋亡。此外,caspase-4还能诱导IL-18的蛋白水解。作为一种固有的炎症性细胞死亡形式,热凋亡有几层调节机制。NLRP3和细胞因子IL-1的表达需要通过TLRs、TNFR或IL-1R的刺激启动,然后NFκB激活。炎症小体传感器通过翻译后修饰,如磷酸化和泛素化(方框中标出)或裂解,受到严重调控。
推荐panel
Luminex:

Reference:
Bertheloot, D., Latz, E., & Franklin, B. S. (2021). Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cellular & molecular immunology, 18(5), 1106–1121. https://doi.org/10.1038/s41423-020-00630-3



 沪公网安备31011502400759号
沪公网安备31011502400759号
 营业执照(三证合一)
营业执照(三证合一)




