








Hello,各位小伙伴九月好呀,夏日的高温终于过去了,迎来了秋天的凉爽宜人。之前很多小伙伴们说想了解单细胞ATAC (scATAC)在植物中的应用,今天给大家介绍一篇在拟南芥中的scATAC和scRNA联用的文章,看看作者运用了scATAC解决了什么问题。
本次介绍的是2024年4月发表于《Advanced Science》的《Multiome in the Same Cell Reveals the Impact of Osmotic Stress on Arabidopsis Root Tip Development at Single-Cell Level》。这篇文章主要讨论了细胞特异性的转录调控网络(TRNs)在植物发育及应对环境胁迫中的重要作用。作者首先提到传统的单细胞单组学技术难以在同一细胞中直接捕捉不同分子层次间的动态关系。尽管一些先进算法可以整合scRNA-seq和scATAC-seq数据,但在细胞类型特异性TRNs的研究中,精确的数据整合仍是挑战。通过同时分析16,670个拟南芥根尖细胞核的基因表达和染色质可及性,重构了控制根尖在渗透胁迫下发育的TRNs。与通常的基于细胞类型的计算整合方法不同,该研究在单细胞水平捕捉了12,968个染色质峰与基因的关联,构建了前所未有精度的TRNs。此外,这些数据更准确地重构了基因表达和染色质状态在细胞状态转变过程中的协同变化。研究发现,在根尖发育中,染色质可及性的变化先于基因表达,表明染色质状态的改变可能为随后的分化步骤做准备。拟时间轨迹分析揭示了渗透胁迫能够改变生毛细胞的功能分化,研究还识别了与胁迫相关的基因和潜在的调控元件,以及胁迫下细胞异质性的增加。
2.1 单细胞内多种组学可生成跨细胞类型的高质量全基因组染色质和表达谱
作者首先从正常条件和渗透胁迫条件下生长的拟南芥根尖中提取细胞核,同时分析转录组(snRNA-seq)和染色质可及性(snATAC-seq)。通过生物学重复实验,研究收集了16,670个细胞核,并获得了高质量的数据。控制组和胁迫组的TSS得分表明数据适用于后续分析。研究分别对snRNA-seq和snATAC-seq数据进行了UMAP降维,并鉴定了15种不同的细胞类型(基于snRNA-seq数据)和7种不同的细胞类型(基于snATAC-seq数据)。这些细胞类型的标记与已知标记基因相符,并且生物学重复样本间的数据一致性较高。通过对单细胞数据的barcod匹配,研究发现snRNA-seq和snATAC-seq数据在大多数细胞类型中高度匹配,表明这些数据的可靠性。
接下来作者讨论了单独使用ATAC-seq注释难以识别罕见细胞类型的问题,例如木质部和韧皮部。尽管标记基因(如QC标记BBM和木质部标记XCP2)的表达在特定细胞类型中有明确限制,使用ATAC-seq注释却无法清晰区分其染色质可及性峰值。通过RNA注释,这种特异性可以被观察到。研究通过分析15种细胞类型中snATAC-seq的基因活性与snRNA-seq的基因表达相关性,发现罕见细胞类型的相关性较低,可能是因为ATAC-seq信号受高丰度细胞的影响,掩盖了来自罕见细胞类型的信号。这强调了在同一细胞中同时进行RNA水平和染色质可及性分析的重要性,能够更全面、准确地表征转录和表观遗传的全貌。由于转录组能更直接反映基因表达水平,因此在后续分析中,研究主要使用RNA注释来识别细胞类型,并通过barcode信息来匹配相应的染色质可及性数据。

图一:同一细胞中的多组学可生成跨细胞类型的高质量全基因组染色质和表达谱。
2.2 染色质可及性和基因表达水平差异标记的细胞状态
接下来作者揭示了染色质可及性和基因表达水平如何在不同细胞类型中标记细胞状态的差异。通过绘制Sankey图,研究展示了RNA注释和ATAC注释之间的相关性,发现大多数初始细胞表现出类似于分化细胞的开放染色质区域,但这些基因的表达水平较低。基于染色质可及性,初始细胞被划分为六个子簇:INIT.表皮/皮层/内皮/中柱/根冠(染色质可及性与分化细胞类似的初始细胞),以及INIT.Pr(染色质可及性类似于原始细胞的初始细胞,如QC和初始细胞)。研究发现,60.95%(控制组)和64.13%(胁迫组)的初始细胞表现出与分化细胞相似的染色质可及性。
细胞比例分析表明,无论是在控制组还是胁迫组,INIT.表皮/皮层/内皮/中柱/根冠簇的比例与相应细胞类型的比例相似。进一步的基因得分和相关性分析显示,INIT.皮层/中柱/内皮与其未来将分化的细胞类型相关性最高,而INIT.Pr与QC、初始细胞及其他原始细胞的相关性较高。此外,INIT.表皮细胞在控制组中与毛根母细胞的相关性最高,而在胁迫组中与表皮细胞的相关性最高,表明控制组的INIT.表皮细胞可能发展为生毛细胞,而胁迫组的INIT.表皮细胞更可能发展为表皮细胞。显微镜观察进一步验证了这一点,发现胁迫条件下生毛细胞数量减少,表明这种现象可以追溯到初始细胞的染色质状态变化,而不仅仅是表皮到生毛细胞的分化过程受到了胁迫的影响。
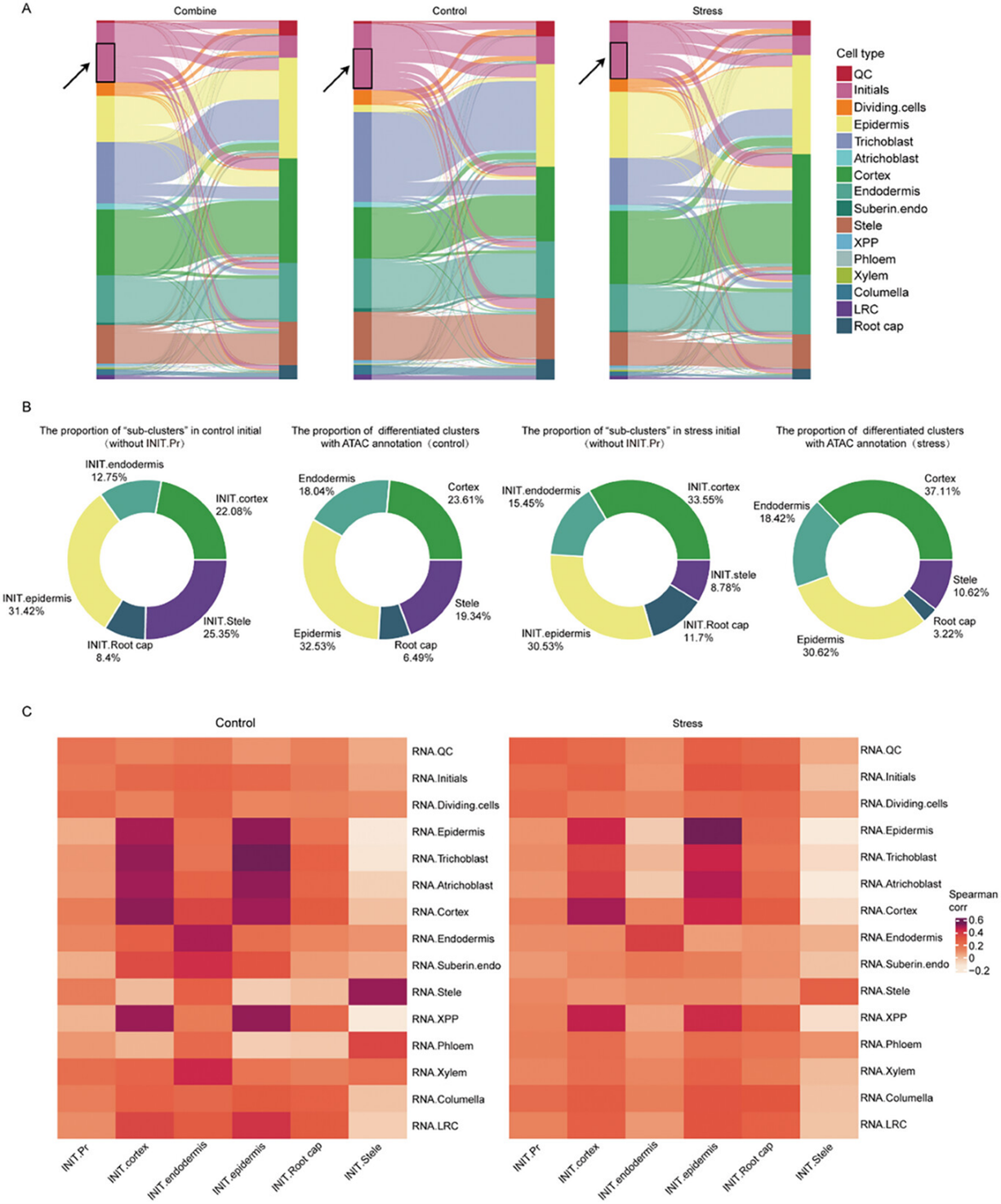
图2:染色质可及性和基因表达水平差异标记的细胞状态
2.3 染色质可及性预示着原代细胞的未来细胞命运
接下来作者使用Monocle2构建了初始细胞的发育轨迹,并基于染色质可及性类型绘制了细胞的分布。发育轨迹显示,INIT.Pr主要集中在轨迹的起点,而那些染色质可及性类似于分化细胞的细胞则位于轨迹的末端(图3A,B)。沿着伪时间顺序将发育轨迹划分为三个阶段,并计算了每个阶段中各“子群”的比例。 INIT.Pr在发育早期的比例非常高,约为75%,并且在中期和后期逐渐减少,同时分化的簇逐渐增加(图3C,D)。然而,初始标记基因的表达水平在整个发育轨迹中并没有显著变化,这表明这些细胞在转录组水平上相似,但在后续的分化过程中,染色质可及性有所不同。为了进一步证实这一推测,作者使用CytoTRACE分析了六个“子群”在对照组和胁迫组中的分化顺序。结果发现,在对照组和胁迫组中,INIT.Pr的分化显著低于其他簇(图3E)。通过独立方法重建的伪时间轨迹结果表明,根尖发育过程中染色质在基因表达启动之前就已经被打开,可能是在为后续的分化步骤做准备,这种现象只能通过在同一个细胞中测量染色质状态和基因表达谱才能被检测到。单核多组学数据的分析为解释细胞类型水平上染色质状态和基因表达之间的不一致提供了新的见解。
在这些提前开放的染色质区域中,作者鉴定出参与磷吸收的基因PHT1;1,该基因在根部外层表皮细胞和一些初始细胞中具有开放的染色质(表现出与表皮相似的染色质可及性)。磷是植物生长和发育的重要宏量营养元素之一。增加磷营养水平可以改善植物适应干旱的能力以及水分利用效率。正如预期的那样,PHT1;1 mRNA在根部最外层表皮细胞中表达,尽管在初始细胞中染色质可及性较高,PHT1;1并没有表达。使用CytoTRACE追踪了发育轨迹中的QC干细胞、初始细胞、表皮细胞、非成毛细胞(atrichoblast)和生毛细胞(trichoblast)。QC细胞位于轨迹的起点,生毛细胞位于轨迹的末端(图3F)。在发育轨迹中同时分析PHT1;1染色质可及性数据和表达水平后,发现在一些初始细胞中,染色质已为转录启动做好准备,但该基因并未表达(图3G,H)。作者还发现,暴露于渗透胁迫的细胞中,PHT1;1的可及性峰值数量和表达水平都高于对照细胞(图3I,J)。
研究还集中分析了与渗透胁迫和生长素相关的基因,发现它们的表达模式和染色质可及性在不同细胞类型中被特异性调节。例如,生长素合成相关基因YUC5在筛管中被抑制,生长素运输相关基因ABCB19和PIN1在根端分生组织中被渗透胁迫特异性抑制。此外,ABA信号通路的关键调控基因ABI5在木质部中被诱导表达。这表明这些基因的激活受细胞类型特定的复杂转录调控控制。
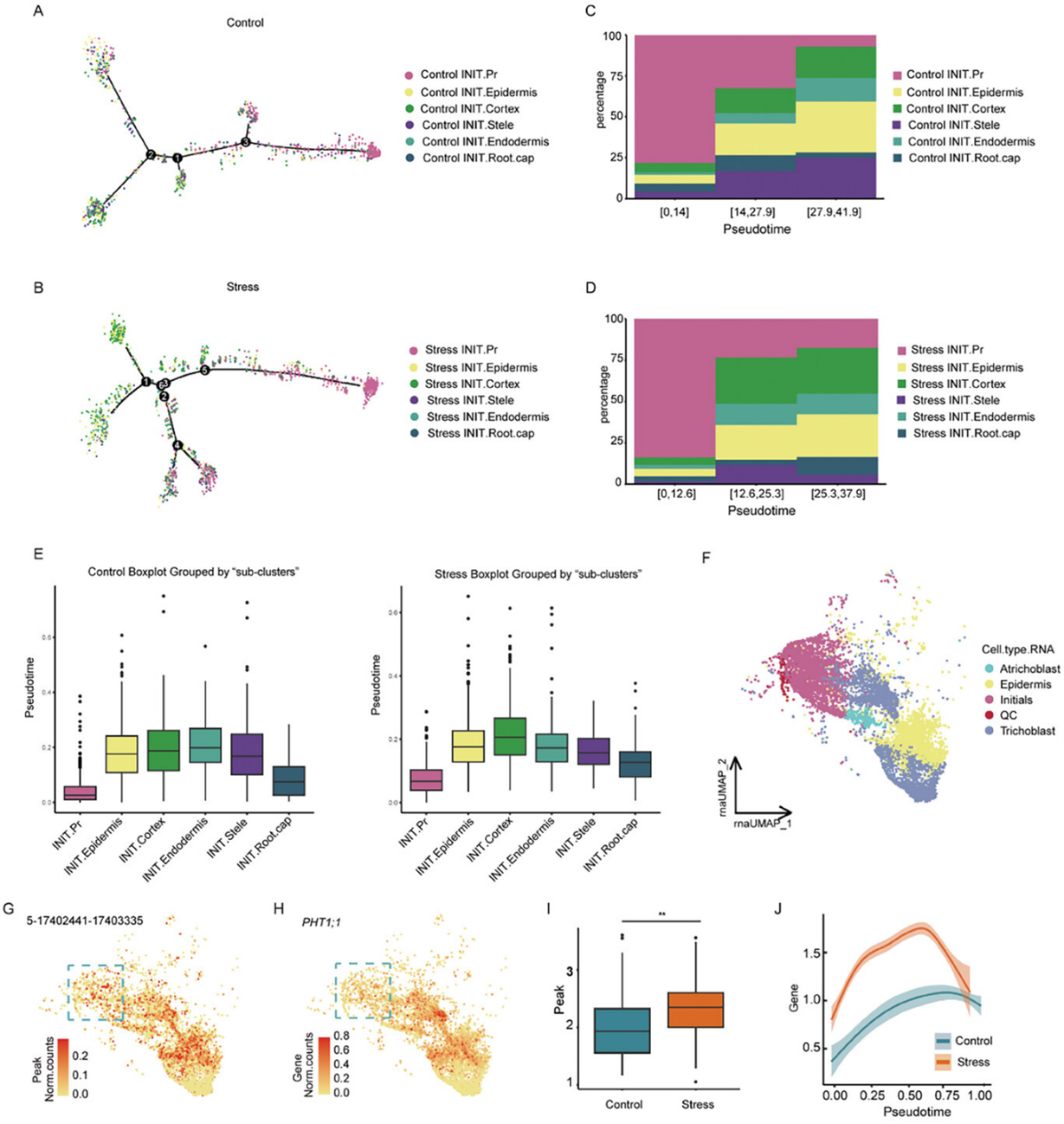
图3:染色质可及性预示着原代细胞未来的细胞命运
2.4 渗透胁迫下细胞类型的特异性变化
渗透胁迫触发了基因表达、发育和植物生理的特定响应。作责首先使用CytoTRACE追踪了从低分化到高度分化的拟南芥根细胞的发育轨迹。结合RNA注释结果,观察到生毛细胞(trichoblast)在尾部富集(表明分化结束),而QC细胞在起点富集(图4A)。对照细胞比受胁迫细胞有更高的拟时间,表明其分化潜力更大(图4B)。这些结果表明,渗透胁迫扰乱了拟南芥根中各个细胞类型的生理功能。差异基因表达分析显示,与韧皮部、Suberin-endo和XPP细胞相比,在生毛细胞、皮层、内皮层和中柱中的差异表达基因更多,尤其是在渗透胁迫下。这些基因包括编码与渗透胁迫相关的蛋白激酶,如MPK3和SnRK2.8,它们在皮层中受到渗透胁迫的高度诱导(支持信息图S5B)。编码Na+转运蛋白的基因HKT1和盐胁迫耐受基因AT1G13930在渗透胁迫下富集于中柱中。还有另一种蛋白激酶SnRK2.7和根向引力相关蛋白SP2L在渗透胁迫下于生毛细胞中被诱导。上述结果表明,这些细胞类型在根尖响应渗透胁迫中可能起到了更为关键的作用。
每个簇种上调基因进行了GO富集分析发现与水分运输和液体运输相关的基因在大多数细胞类型中表达水平升高,除了生毛细胞、皮层、内皮层和中柱外。皮层、内皮层和中柱细胞对渗透胁迫有类似的反应,渗透胁迫诱导了与前体代谢物生成和ATP代谢过程相关的基因在这些细胞中的上调。有趣的是,生毛细胞对渗透胁迫表现出特定的响应模式,富集于与有氧呼吸、细胞呼吸和氧化磷酸化相关的途径中,表明在每种细胞类型中可能存在应对外界环境因素的不同机制。生毛细胞(根毛)是根尖中吸收水分、无机盐和感知环境变化的重要细胞类型,与其他细胞类型相比,生毛细胞在渗透胁迫下有最多的差异表达基因。同时还发现,与对照组相比,胁迫样品中的生毛细胞减少。CytoTRACE结果显示,在对照组中生毛细胞的分化高于胁迫组(图4C),与总体水平结果一致(图4B)。随后通过重构对照和胁迫细胞的拟时间轨迹分析了渗透胁迫对生毛细胞发育的影响。使用Monocle2进行生毛细胞和QC细胞的拟时间分析,并定义QC为拟时间轨迹起点位置(图4D-F)。在起点附近,胁迫组和对照组沿相同轨迹分叉,在分叉点1形成两个分支:分支1和分支2。 研究进一步发现,基于轨迹中的基因表达变化,可以将基因分为三个簇(图4G)。簇1中的基因在分支2中高表达,主要调控分支2的发育,这些基因与缺氧响应、铁运输和细胞内铁平衡相关,且在对照组中占了很大比例(图4G)。在分支1的发育轨迹中,主要在胁迫组中的生毛细胞中表达了与表皮分化、生毛细胞发育和水分运输相关的基因(簇2和簇3)(图4G)。拟时间轨迹重建表明,拟南芥的生毛细胞有两种亚型,具有不同的基因表达谱:其中一种亚型为成熟的生毛细胞,主要响应环境变化和离子吸收(分支2;图4F);另一种为分化中的生毛细胞(分支1;图4F),大多数经过验证的生毛细胞标记基因在此处表达。上述结果表明,渗透胁迫改变了生毛细胞的生理状态,并抑制了一部分生毛细胞从分化到成熟的转变,从而影响了其生理功能(图4D-F)。
接下来作者使用SCENIC分析了拟南芥TF家族在生毛细胞中的表达,并绘制了它们的调控子活性热图和调控网络(图4H、I)。分析发现,WRKY家族的成员WRKY17(AT2G24570),一种由非生物胁迫(盐、甘露醇、干旱)诱导的植物特异性TF,在渗透胁迫下在生毛细胞中高表达(图4H)。与此一致,WRKY17的基序可及性在生毛细胞中增加。更重要的是,进一步观察到那些启动子含有WRKY17结合基序的靶基因在胁迫组中上调。在生毛细胞调控网络中,作者还鉴定出了WRKY11(AT4G31550),其在系统发育上与WRKY17相关,且也受渗透胁迫诱导(图4H)。值得注意的是,WRKY11的基序可及性和靶基因表达模式与WRKY17相似。通过qPCR检测,观察到WRKY17和WRKY11在胁迫处理的拟南芥根尖中的表达水平较对照组高。WRKY转录因子在调控植物对生物和非生物胁迫的响应中起着重要作用,尽管在多种植物物种中进行了研究,但WRKY转录因子的细胞类型特异性响应仍未完全解析。通过单细胞多组学技术,发现WRKY11/17在渗透胁迫下在生毛细胞中高表达,可能在渗透胁迫响应中发挥关键作用。
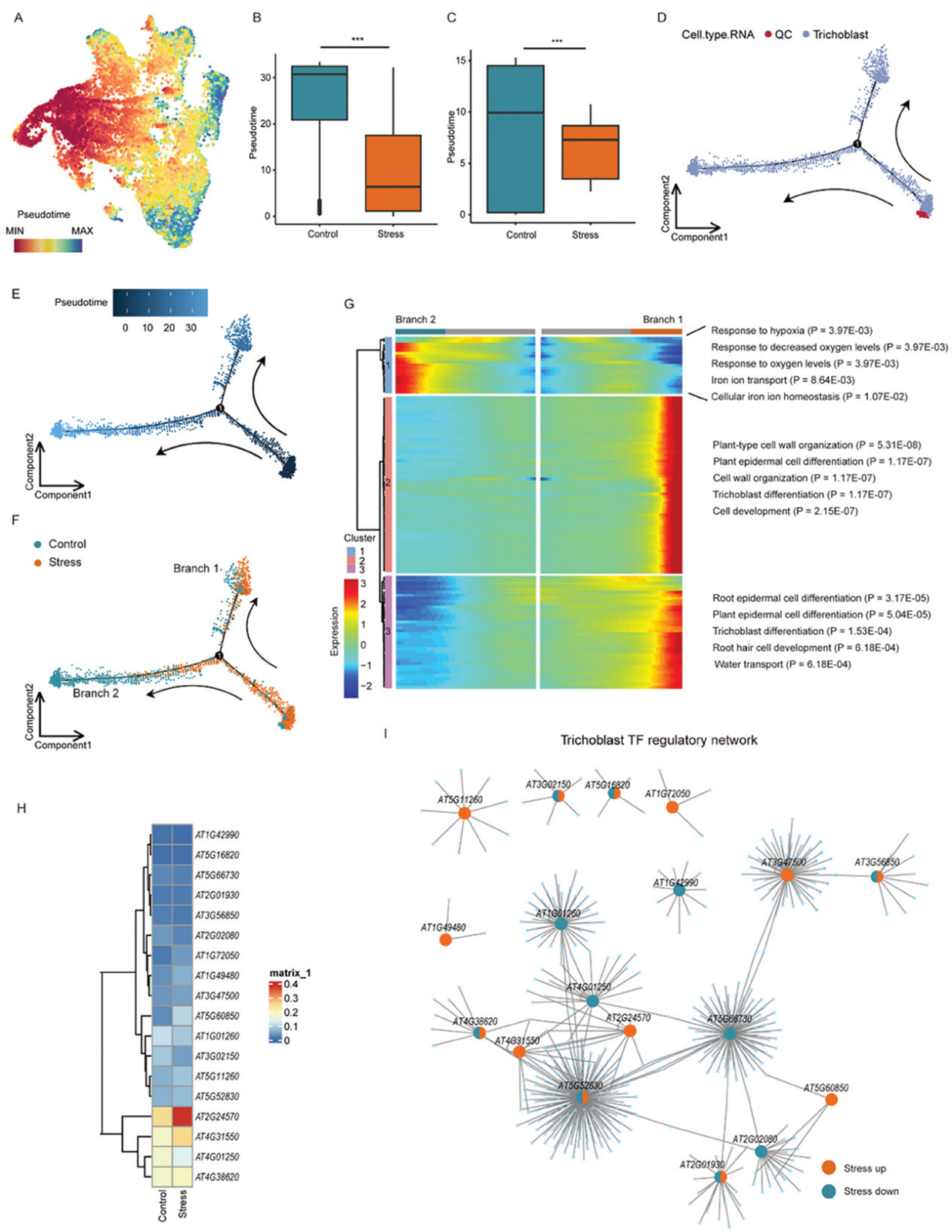
图4:渗透压下细胞类型的特异性变化
2.5 渗透胁迫下的细胞类型特异性顺式调控
植物中的基因表达由顺式调控元件(CREs)调节,例如启动子和增强子,这些元件响应发育和环境变,使用单细胞多组学技术可以更深入地理解细胞类型特异性的基因调控机制。通过在同一细胞中使用单细胞多组学ATAC+基因表达技术,能够以更高的精度识别细胞特异性基因相关的候选顺式调控元件(gl-cCREs)。通过分析snATAC-seq数据,检测共可及性峰对预测基因组中的顺式调控相互作用,其中一个峰位于启动子区域,且染色质可及性与下游基因表达呈正相关。基于此预测了一组CRE启动子对,并在伪散样(pseudo-bulk)中鉴定出7,373个gl-cCREs和3,763个cCRE相关基因。因为gl-cCREs与已验证的拟南芥根部增强子序列重叠,且gl-cCREs包含超级增强子(SE)序列,证明了分析的可靠性。
随后,使用snATAC-seq数据预测了不同细胞类型中与胁迫相关的gl-cCREs,发现基于折叠变化直方图,胁迫相关gl-cCREs的数量远高于伪散样,其中根冠胁迫样本中的gl-cCREs数量约为伪散样的十倍(图5A)。这些结果表明,单细胞多组学ATAC + 基因表达能够获得更多关于应激相关cCRE的信息,因为一些细胞特异性的峰值在细胞类型水平整合snRNA-seq和sATAC-seq数据时会被淹没,从而无法识别。接着,作者重点分析了胁迫相关的gl-cCREs,其中与其他峰对相比有更多的相互作用峰对,这些被定义为胁迫相关的热点gl-cCREs(hgl-CREs),总共发现了240个hgl-cCREs(图5B-F)。以前的研究表明,增强子在不同物种中具有相似的功能,并驱动发育基因的细胞特异性表达。为了探讨发现的hgl-cCREs的保守性,作者分别将gl-cCREs和hgl-cCREs的序列与已发表的十字花科家族的保守非编码序列进行了比较。作者发现,大多数hgl-cCREs包含多个保守非编码序列,且含有这些序列的hgl-cCREs的比例显著高于gl-cCREs(图5G)。这些结果表明,一些保守非编码序列可能作为增强子元件来调控拟南芥在渗透胁迫下多个基因的表达(图5H),这些序列可能在其他十字花科物种中发挥类似作用,如芜菁和阿拉伯蕨苔。
鉴于根冠中胁迫相关gl-cCREs和靶基因的数量显著高于其他细胞类型(图5A、I),作者对胁迫相关gl-cCREs的靶基因进行了富集分析。富集分析表明,这些基因与组蛋白H3K4甲基化相关,或者是组蛋白H3K4甲基化复合物的核心成分,或参与调控组蛋白H3K4甲基化及其他重要生物过程(图5J)。顺式调控元件分析表明,胁迫相关gl-cCREs主要参与激活根冠细胞对渗透胁迫的H3K4甲基化相关基因表达。同时,作者检测了在渗透胁迫下组蛋白甲基化的变化,使用CUT&Tag技术检测了对照组和胁迫组的H3K4me3整体水平。测序数据显示,生物学重复样本是一致且可靠的,且在渗透胁迫下H3K4me3修饰水平显著增加。H3K4me3修饰基因的富集分析显示,H3K4me3相关基因参与了氧化磷酸化、铁运输、茉莉酸合成和损伤响应,在渗透胁迫下上调根冠特异性顺式调控元件和H3K4修饰之间可能确实存在调控关系,但其确切机制仍需要进一步详细研究。
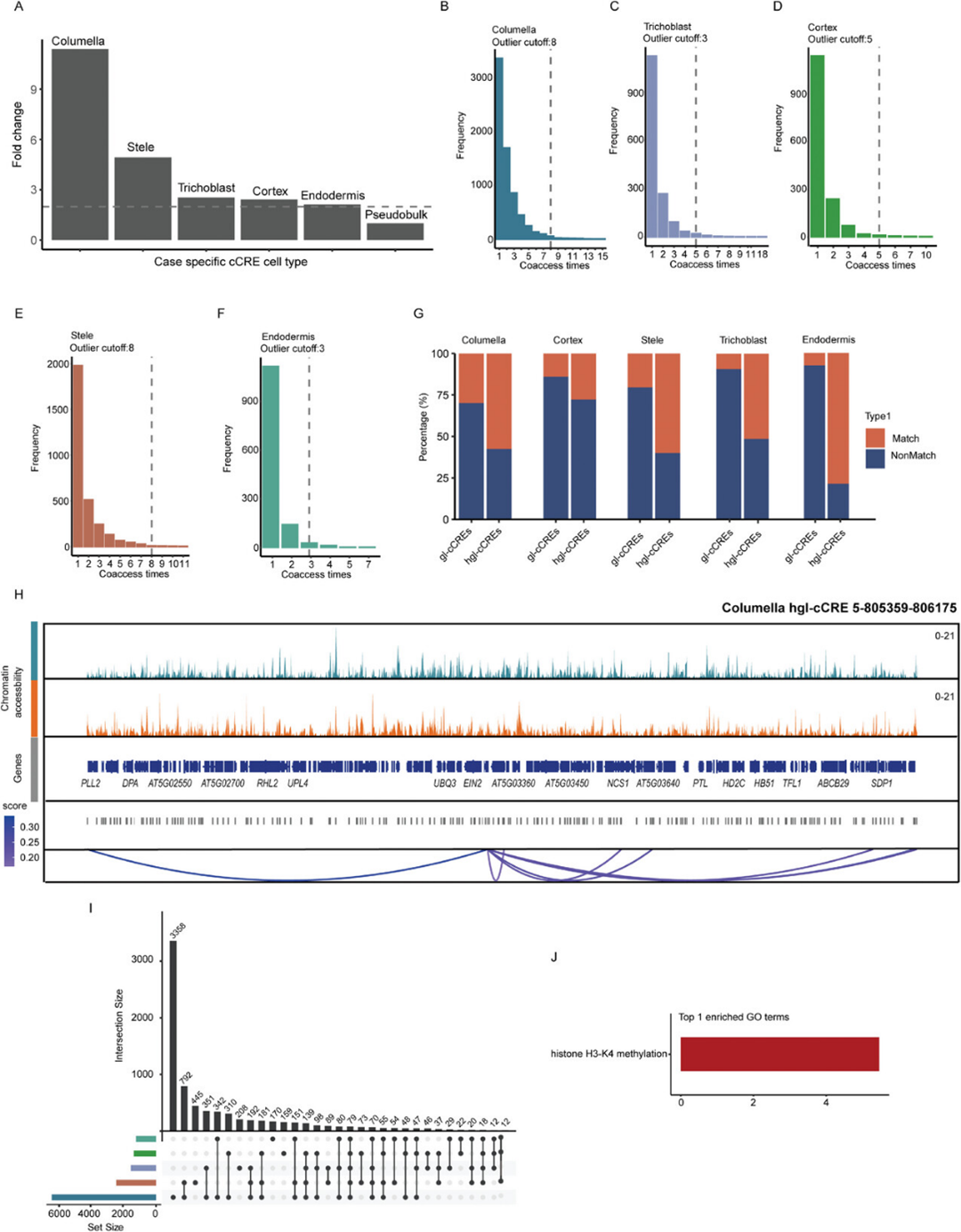
图5:渗透胁迫期间细胞类型特异性转录顺式调节
结语:
以上就是文章的整体内容了,可以看到基于单细胞ATAC结果的分辨率不足,结合单细胞转录组既可以提高分辨率,进而解析不同细胞类型下的调控网络差异。乐备实生信部在单细胞多组学研究中具有丰富的经验,并且我们引入了稳定强大的BD Rhapsody ATAC-Seq平台,助力客户表观调控研究。欢迎联系合作。



 沪公网安备31011502400759号
沪公网安备31011502400759号
 营业执照(三证合一)
营业执照(三证合一)




